
Transforming growth factor-β(TGF-β) is a pro-metastatic factor in the late-stage breast cancer. In addition to the canonical SMAD pathway, TGF-β receptors can initiate other intracellular pathways via either phosphorylation or direct interaction with signaling intermediates; these so-called non-SMAD signaling pathways include several branches that involve phosphatidylinositol kinase (PI3K)/AKT, mitogen-activated protein kinases (MAPKs), and Rho-like GTPase signaling intermediates.
Oncogenic PI3K/AKT signaling antagonizes TGF-β-induced growth arrest and apoptotic responses. Moreover, high TGF-β levels in tumour correlate with overactive PI(3)K�AKT signalling, and poor prognosis in breast cancer. However, how AKT cross-reacts with TGF-β-induced pro-invasive and pro-metastatic responses in advanced tumors remains undefined.
In the TGF-β/SMAD canonical pathway, TβRI acts downstream of TβRII; the stability and membrane localization of TβRII are therefore critical determinants of both the sensitivity and duration of the TGF-β response. Many previous studies have concluded that TβRII mediates cytostatic effects of TGF-β; loss of its function in many different cancer models promotes aggressive and metastatic behavior. Whether a gain of function in TβRII can promote metastasish as not been thoroughly investigated. And the regulation of TGF-β type II receptor remains uncertain.
In this work, we identify FAF1 as a key regulator of cell surface TβRII. And FAF1 can destabilize TβRII on the cell surface by recruiting VCP/E3 ligase complex thereby limiting excessive TGF-β response. During cancer progression, growth factor-induced (or oncogenic mutation) activation of AKT directly phosphorylates FAF1 at Ser 582, which disrupts the FAF1-VCP complex and reduces FAF1 at the plasma membrane. The latter results in an increase in TβRII at the cell surface that promotes both TGF-β-induced SMAD and non-SMAD signaling.
We uncover metastasis suppressing role for FAF1 through analyses of FAF1-knockout animals, various in vitro and in vivo models of epithelial to mesenchymal transition and metastasis, an MMTV-PyMT transgenic model of mammary tumor progression and clinical breast cancer samples. These findings uncover a previously uncharacterized mechanism by which TβRII is tightly controlled. Together, we reveal how SMAD and AKT pathways interact to confer pro-oncogenic responses to TGF-β.
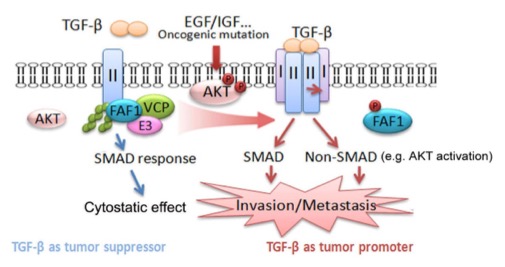
Figure: AKT antagonizes FAF1 tomaintain TβRII stability in metastaticbreast cancer.
The first authors of this article are Feng Xie, Ke Jin and Li Shao. The corresponding author is Professor Long Zhang in Life Sciences Institute, Zhejiang University.
Text link: https://www.nature.com/articles/ncomms15021

