

2024年3月27日,我院蒋超实验室在生物信息学领域著名期刊Briefings In Bioinformatics 上在线发表了题为“Generalized reporter score-based enrichment analysis for omics data”的研究论文。该工作开发了面向多组学及纵向组学数据富集分析的Generalized Reporter Score-based Analysis(GRSA)方法及对应R软件包。
功能富集分析是一种主流的生物信息学分析方法,有助于了解大型组学数据集(如转录组、宏基因组和代谢组数据)的生物学意义。我们可以通过识别丰富的功能类别(例如GO或KEGG pathway)来深入了解潜在的生物学过程和功能,并为下游实验提供假设。然而,大多数现有工具无法直接将富集分析应用于多群组(Multi-group)和纵向(Longitudinal)组学数据。基于Reporter Score的富集分析(RSA)方法显示出卓越的敏感性,因为它依赖于p值而不是特征原始值,但由于缺乏适当的工具经常被误用。
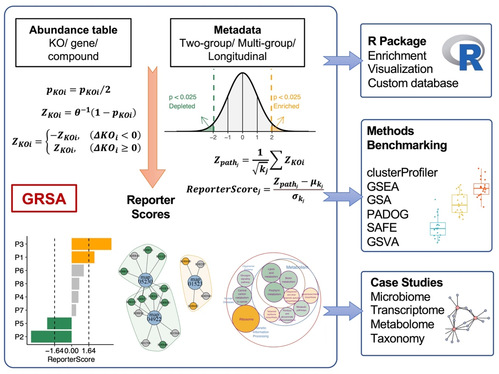
研究团队开发了改进的基于广义Reporter Score的分析方法(GRSA),该方法可在R包 ReporterScore中实现(https://github.com/Asa12138/ReporterScore),并提供了全面的可视化方法和通路数据库。GRSA 是一种无阈值富集方法,适用于所有类型的生物医学特征,例如基因、化合物和微生物物种等。GRSA支持多群组和纵向实验设计,因为其包含与其兼容的统计方法。ReporterScore包还支持自定义分层和关系数据库,为高级用户提供了额外的灵活性。
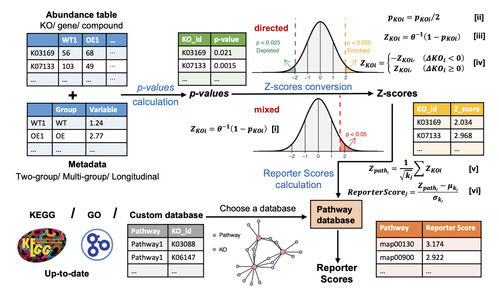
GRSA的工作流程
研究团队在多个数据集中对 GRSA 与其他流行的富集方法(clusterProfiler,GSEA,GSA,PADOG等)进行了基准测试。GRSA显示出比其他常用富集分析方法更高的优先度和灵敏度,可以有潜力地识别出与所研究疾病相关的更多生物学途径。研究团队在4个有代表性的组学研究案例中使用了GRSA(1. 皮肤微生物组的年龄相关功能动态分析;2. 心肌细胞分化过程中的转录组动态变化;3. 胎龄相关的母体代谢组系统性变化;4. 皮肤微生物组的Taxonomy富集分析)。GRSA结果不仅确认了之前的主要发现,并针对不同数据揭示了新的生物学见解,展示了GRSA在各类实验设计(两组,多群组,纵向)及各类组学数据中的有效应用。
总之,该研究开发的GRSA方法和ReporterScore软件包可以极大地促进不同组学数据的功能富集分析,并具有更高的灵敏度,与多组和纵向设计的兼容性,以及可自定义数据库的灵活性。研究团队相信,GRSA和ReporterScore软件包将在生物医学研究领域的各类组学数据中有广泛的应用。
蒋超实验室博士生彭晨为论文的第一作者,蒋超研究员为论文的通讯作者,参与该研究的还有蒋超实验室博士后陈琼,华大研究员谭上进和斯坦福大学博士后申小涛。该研究得到了国家自然科学基金等项目资助,以及生命科学研究院NECHO高性能计算集群的支持。
原文链接:https://doi.org/10.1093/bib/bbae116

